C’est un cas clinique digne d’un épisode de la série Dr House que des médecins du Massachusetts General Hospital de Boston ont rapporté dans un article publié en ligne le 3 avril 2024 dans l’hebdomadaire médical américain The New England Journal of Medicine. C’est l’histoire d’une femme noire de 46 ans, diabétique de type 2, hospitalisée pour une hyperglycémie persistante alors qu’elle reçoit quotidiennement de l’insuline à très forte dose, à raison de 2 000 unités par jour (2 000 U/j).
Voilà cinq ans qu’un diabète de type 2 a été diagnostiqué chez cette quadragénaire qui se plaignait de nausées, vomissements, polyurie et polydipsie. Elle pesait alors 113 kg et son IMC (indice de masse corporelle) était de 45,6. Un traitement par metformine et insuline avait été commencé à la dose de 10 unités par jour. La metformine avait dû être arrêtée du fait de la survenue d’effets secondaires indésirables et remplacée par le glipizide, un sulfamide hypoglycémiant. Durant les trois ans et demi qui ont suivi, le contrôle glycémique s’est détérioré malgré une perte de poids. Le traitement hypoglycémiant avait alors été progressivement augmenté.
Il y a deux ans, cette patiente a déménagé dans la région de Boston et a été prise en charge dans un hôpital. Son poids est alors de 63 kg et son IMC à 25,4. Elle n’est donc plus obèse, seulement en très léger surpoids. Elle est traitée par insuline (20 U/j) et glipizide. Son hémoglobine glyquée est à 13,3 % (pour une valeur normale de 5,6 %). La dose d’insuline glargine par voie sous-cutanée est augmentée à 30 U/j, en même temps que de l’insuline lispro est ajoutée à raison de 10 U trois fois par jour avec les repas. Alors que le glipizide est arrêté, la patiente reçoit en plus deux autres antidiabétiques oraux (glimépiride et pioglitazone).
Il y a un an, son poids est descendu à 48 kg et son IMC à 19,4, donc normal. Son traitement consiste en de l’insuline glargine (34 U/j), de l’insuline lispro sous-cutanée (15 U trois fois par jour avec les repas), du glimepiride et de la pioglitazone. L’hémoglobine glyquée est alors pourtant à 14 %. La patiente est anorexique et fatiguée. Alors qu’elle mange peu, sa glycémie reste élevée.
Il y a neuf mois, son poids était de 42 kg et son IMC à 16,9. Elle recevait alors de l’insuline glargine (46 U/j), de l’insuline lispro sous-cutanée (20 U trois fois par jour avec les repas), du glimépiride, et de la pioglitazone. L’hémoglobine glyquée était à 11 %.
Il y a six mois, la patiente s’est plainte de douleurs abdominales persistantes qui ont été mises sur le compte d’une gastroparésie, trouble fonctionnel digestif défini par un ralentissement de la vidange de l’estomac en l’absence de tout obstacle mécanique. L’hémoglobine glyquée était à 15,5 %.
Il y a deux mois, les douleurs abdominales ont augmenté d’intensité et la patiente a de nouveau été hospitalisée, dans un autre hôpital. La dose d’insuline a alors été portée à 500 U au petit-déjeuner et au déjeuner et à 450 U au dîner. En plus, une insuline de durée d’action intermédiaire (insuline isophane ou NPH) a été ajoutée à la dose de 80 U deux fois par jour. Les douleurs abdominales ont disparu en même temps que le contrôle glycémique s’est amélioré. La patiente a pu regagner son domicile.
Au cours des deux mois suivants, la patiente a été hospitalisée à cinq reprises pour douleurs abdominales et hyperglycémie. À chaque admission, de l’insuline par voie intraveineuse a été administrée. Les douleurs ont disparu et cette femme a pu quitter l’hôpital.
Il y a neuf jours, les douleurs abdominales se sont aggravées. La patiente a dû être transportée en ambulance aux urgences d’un hôpital. Là encore, un traitement par insuline a été initié. Le lendemain, l’insulinothérapie intraveineuse a été interrompue et l’insuline a été prescrite à raison de 650 U à chaque petit-déjeuner et déjeuner, avec en plus 600 U au dîner. Un traitement par insuline NPH a également été prescrit à raison de 80 U deux fois par jour.
C’est alors que cette patiente est transférée au Massachusetts General Hospital. À son arrivée dans ce nouvel hôpital, la patiente se plaint de douleurs abdominales constantes. Elle indique s’administrer de l’insuline, sans jamais oublier de doses et précise qu’elle effectue, comme il se doit, une rotation des points d’injection à l’intérieur d’une même région anatomique. Elle est dépressive. Elle présente par ailleurs, depuis neuf mois, un engourdissement des mains et des pieds, associée à des picotements. Elle n’a plus ses règles depuis 14 mois, probablement du fait d’un faible IMC.
Cette femme vit alors à Boston avec son mari et ses enfants. Auparavant, elle occupait un emploi de chef dans un restaurant, mais ne travaille plus actuellement. Elle ne boit pas d’alcool, n’a jamais fumé et ne consomme pas de drogues illicites. Sa mère avait un diabète de type 2.
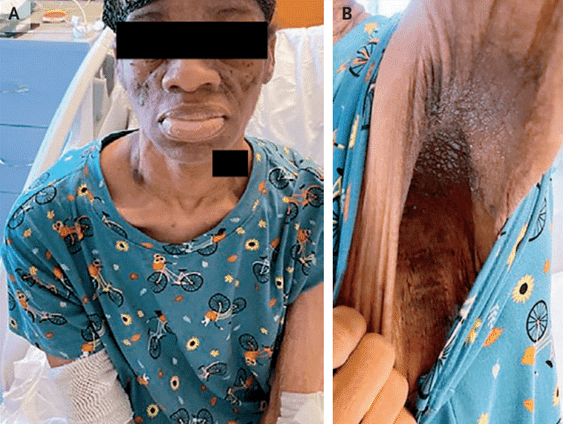
Son poids est maintenant de 42 kg et son IMC est descendu à 17. Elle a beaucoup perdu de graisse corporelle et de masse musculaire.
L’examen clinique trouve un visage présentant des traits ressemblant à une acromégalie, ce que l’on appelle un faciès acromégaloïde, en l’occurrence un élargissement du nez et un épaississement des lèvres. De plus, cette femme présente sur le cou et dans les aisselles des lésions hyperkératosiques et pigmentées de l’épiderme, donnant à la peau un aspect rugueux, épais, et une couleur brunâtre, ce que l’on appelle un acanthosis nigricans. Le foie et la rate sont de volume normal. La peau des jambes est abimée.
La glycémie à jeun est à 3,11 g/dL (pour une valeur normale comprise entre 0,7 et 1 g/dL). Le taux de peptide-C, qui renseigne sur la production endogène d’insuline, est de 12,1 ng/mL (valeurs normales : 1,1-4,4 ng/mL). Les taux de cholestérol et de triglycérides sont normaux, de même que celui des enzymes hépatiques. Le taux sanguin de leptine est indétectable. Le dosage sanguin de l’adiponectine est de 24,5 µg/mL (valeurs normales : 2,9-30,4). La sérologie VIH est négative. Le dosage de certains auto-anticorps (anticorps antinucléaires) est positif avec un titre de 1:320, ce qui témoigne d’une anomalie du système immunitaire (trouble dysimmunitaire).
Le scanner abdomino-pelvien sans produit de contraste révèle l’absence quasi-totale de graisse viscérale et intra-abdominale. De nombreux plis cutanés témoignent de cette absence significative de tissu adipeux. Le pancréas est atrophique, ce que l’on observe fréquemment chez les sujets ayant un diabète chronique insulino-dépendant.
Résumons. Nous sommes en présence d’une femme à qui on a diagnostiqué un diabète de type 2 (DT2) cinq ans plus tôt et chez qui les doses d’insuline exogène nécessaires pour réduire sa glycémie ont progressivement augmenté, traduisant une résistance à l’insuline, c’est-à-dire une diminution de la réponse biologique à cette hormone hypoglycémiante. Mais de quoi souffre donc cette femme de 46 ans ?
Comme le rappellent Lindsay Fourman, Stephen O’Rahilly et ses collègues du Massachusetts General Hospital, « la sécrétion physiologique d’insuline est de l’ordre de 0,4 à 1 unité par kilogramme de poids corporel par jour. Des besoins en insuline exogène supérieurs à 2 ou 3 unités par kg et par jour témoignent d’une résistance sévère à l’insuline. Or, au moment de son transfert dans cet l’hôpital, cette patiente avait un besoin d’insuline étonnamment élevé, dépassant les 50 unités par kg et par jour ».
Avant de conclure qu’un patient présente une résistance à l’insuline, il importe de s’assurer qu’il suit bien son traitement, ne consomme pas trop de glucides et que l’insuline sous-cutanée n’est pas injectée dans une zone cicatricielle ou œdémateuse, ce qui peut réduire la diffusion de l’insuline dans les tissus. Enfin, il convient que l’insuline ne soit pas périmée ou conservée dans des conditions inadéquates (au soleil ou en dehors du réfrigérateur). Dans la mesure où la patiente a reçu, alors qu’elle était hospitalisée, à la fois de l’insuline par voie intraveineuse et sous-cutanée, la possibilité qu’il ne s’agisse pas d’une véritable insulino-résistance est improbable. Elle présente par ailleurs les signes cliniques d’une insulino-résistance (acanthosis nigricans, faciès acromégaloïde, lésions cutanées) qui surviennent du fait que l’insuline en excès stimule le récepteur de l’IGF-1 (insulin growth factor-1). On rappelle que le taux d’IGF-1 est élevé dans l’acromégalie.
Chez cette patiente, l’obésité, observée au moment où son diabète a été diagnostiqué, ne peut expliquer son état clinique. En effet, ses besoins en insuline n’ont pas cessé d’augmenter alors même qu’elle perdait du poids. Ils étaient même au plus haut alors qu’elle présentait une extrême maigreur.
On sait que certains médicaments peuvent diminuer la réponse biologique à l’insuline. C’est le cas des glucocorticoïdes, des antipsychotiques, des antirétroviraux en cas d’infection par le VIH. Mais la patiente n’a reçu aucun de ces traitements.
Diagnostics différentiels
Les médecins vont alors évoquer différents diagnostics différentiels, c’est-à-dire l’ensemble des pathologies dans lesquelles on observe une insulino-résistance.
Et de penser au glucogonome, tumeur maligne, plus rarement bénigne, développée à partir des cellules alpha des îlots pancréatiques qui sécrètent une hormone hyperglycémiante : le glucagon. Mais la patiente ne présente pas le signe clinique capital du glucagonome : l’érythème nécrolytique migrateur. Ce signe caractéristique correspond à des macules, papules et bulles qui évoluent vers l’ulcération. Ces lésions de couleur rouge sombre, volontiers érosives, à évolution centrifuge, touchent de façon privilégiée les extrémités des membres, la région autour de la bouche et la région périgénitale.
La patiente serait-elle atteinte du syndrome de Cushing, caractérisé par des signes cliniques associés à une exposition prolongée à des taux élevés de cortisol circulant ? Là encore, les signes cliniques typiques de cette pathologie endocrinienne (prise de poids associée à une obésité tronculaire, faciès lunaire, tendance aux ecchymoses, stries violacées, amyotrophie des jambes et des bras) ne sont pas présents.
Et si la patiente souffrait d’acromégalie, pathologie résultant d’une sécrétion excessive d’hormone de croissance et presque toujours due à un adénome hypophysaire (tumeur bénigne de l’hypophyse) ? Elle présente bien un des traits acromégaloïdes du visage au niveau du nez et des lèvres, mais pas d’épaississement des os à l’extrémité des doigts, pas de douleurs articulaires, pas d’œdème des tissus mous autour des os, autant de signes caractéristiques de l’acromégalie. De plus, le taux d’IGF-1 n’est pas élevé.
Une résistance à l’insuline peut se développer chez des patients présentant une lipodystrophie, caractérisée par une distribution anormale des masses adipeuses de l’organisme. Mais dans cette affection, la masse musculaire est préservée, alors que la patiente présente une atrophie musculaire et peu de graisse corporelle, ce qui traduit une maigreur extrême (cachexie). Cependant, le diagnostic de lipodystrophie ne peut pas être totalement exclu car une absence de graisse sous-cutanée associée à un faible taux de leptine circulante peut se voir dans cette affection et dans la cachexie. Le diagnostic de lipodystrophie reste donc toujours possible.
L’insuline joue un rôle anabolique majeur, favorisant la mise en réserve du glucose dans le foie et les muscles. Ceci explique qu’une résistance à l’insuline conduise à la fonte (catabolisme) du tissu musculaire et adipeux. Les effets de l’insuline résultent de sa liaison à un récepteur membranaire spécifique exprimé en priorité sur ses trois tissus cibles que sont le foie, le muscle et le tissu adipeux.
Un autre diagnostic reste donc à envisager, celui d’une résistance à l’insuline qui serait liée à l’impossibilité de celle-ci d’agir sur son récepteur, soit du fait qu’elle serait bloquée avant de l’atteindre, soit qu’elle ne pourrait pas s’y fixer, soit qu’elle ne produirait pas son action après liaison à son récepteur. En d’autres termes, le problème serait situé en amont, sur, ou en aval du récepteur de l’insuline.
Les anomalies situées en aval du récepteur à l’insuline sont causées par des anticorps anti-insuline qui se lient à l’hormone circulante. Dans un tel cas, on n’observe généralement pas d’acanthosis nigricans car l’insuline ne peut plus se lier au récepteur de l’IGF-1. Or, la patiente présente un acanthosis nigricans, ce qui n’est donc pas en faveur du diagnostic d’une insulino-résistance imputable à une anomalie en amont du récepteur de l’insuline.
L’anomalie moléculaire à l’origine de la pathologie de cette patiente se situerait-elle au niveau du récepteur de l’insuline, soit parce que causée par une mutation dans le récepteur ou du fait de la présence d’anticorps dirigés contre ce récepteur ? Une mutation présente sur une copie (mutation hétérozygote) ou les deux copies (mutation hétérozygote) du gène codant le récepteur de l’insuline peut entraîner un syndrome de résistance à l’insuline dit de type A, pathologie qui se manifeste cliniquement pendant l’adolescence ou après.
Une autre maladie génétique rare appartenant au groupe des syndromes d’insulino-résistance extrême, dénommée syndrome de résistance à l’insuline de type B, est due à la présence d’auto-anticorps dirigés contre le récepteur de l’insuline. Ce trouble touche principalement les adultes d’âge moyen, avec une prédominance chez la femme. Il survient par ailleurs plus fréquemment chez les femmes noires qui présentent par ailleurs une maladie auto-immune bien caractérisée ou un trouble du système immunitaire.
Le syndrome de résistance à l’insuline de type B se manifeste en général par l’apparition et le développement rapide d’un diabète très insulino-résistant, accompagné d’acanthosis nigricans, d’une perte de poids chez les femmes. On observe parfois des hypoglycémies, même très sévères, qui peuvent être, dans de rares cas, la seule manifestation métabolique de la maladie.
Quant aux défauts moléculaires situés en aval du récepteur de l’insuline, ils ne se rencontrent que chez des patients présentant une obésité ou une lipodystrophie.
Parce que la patiente est une femme, afro-américaine de surcroît, et qu’elle présente un trouble dysimmunitaire, le diagnostic d’un syndrome de résistance à l’insuline de type B, dû à la présence d’anticorps anti-récepteur de l’insuline, est probable. Les médecins tiennent enfin une piste solide, même si, à ce stade, celle de la lipodystrophie ne peut être totalement écartée.
Syndrome d’insulino-résistance de type B
Le diagnostic du syndrome de résistance à l’insuline de type B repose sur le tableau clinique et biologique. Ce trouble s’accompagne notamment d’une teneur normale en graisse hépatique, d’un taux plasmatique de triglycérides normal et d’un taux sanguin d’adiponectine élevé ou proche des valeurs supérieures de la normale, ce qui est le cas chez cette patiente.
Tout semble donc aller dans le sens du diagnostic du syndrome de résistance à l’insuline de type B. Exit le diagnostic présumé de lipodystrophie !
Ce syndrome d’insulino-résistance extrême, d’origine auto-immune, est rare, mais traitable. Il a été rapporté pour la première fois en 1976 dans la littérature médicale. Reste alors, pour le confirmer, à détecter des auto-anticorps anti-récepteurs de l’insuline dans le sérum. Mais cette détection spécifique n’est que rarement proposée en routine diagnostique, le diagnostic reposant le plus souvent sur des arguments de présomption.
Cela dit, les médecins du Massachusetts General Hospital de Boston ont voulu en avoir le cœur net d’autant plus qu’ils disposent d’un nouveau test de détection de ces anticorps, développé en 2023. Celui-ci s’est révélé fortement positif.
Comme le précisent les auteurs de ce cas clinique, le but du traitement du syndrome de résistance à l’insuline de type B est la rémission complète du diabète résultant de l’élimination de l’anticorps dirigé contre le récepteur de l’insuline. Une rémission spontanée peut se produire chez certains patients, mais le temps nécessaire à la rémission peut prendre plusieurs années. Et d’ajouter qu’« en l’absence de traitement, le syndrome de résistance à l’insuline de type B est associé à un taux de mortalité proche de 50 %. En raison de la rareté de ce trouble, on manque de données issues d’essais randomisés et contrôlés évaluant des traitements immunosuppresseurs ou entraînant l’élimination des anticorps. Plusieurs approches thérapeutiques ont été testées chez un petit nombre de patients, notamment la plasmaphérèse, les échanges plasmatiques, des immunoglobulines intraveineuses et des agents immunosuppresseurs (rituximab, glucocorticoïdes, azathioprine, cyclophosphamide, mycophénolate mofétil) ».
Parce que la rémission peut prendre de longs mois, le traitement efficace du diabète est une priorité. « Le but est d’inverser le processus catabolique [à l’origine de la perte de tissu musculaire et cutané] que l’on observe chez la plupart des patients, et leur permettre d’être traités en sécurité par des immunosuppresseurs, tout en réduisant à la fois le risque d’acidocétose diabétique et d’hypoglycémie », soulignent les auteurs.
La dose médiane d’insuline utilisée dans une étude américano-britannique, publiée dans Diabetes Care en 2018 et portant sur 22 patients, était de 1 775 unités par jour. Une rémission avait été obtenue chez 19 des 22 patients après cinq mois de traitement. Elle avait été objectivée par l’arrêt de l’insuline chez tous ces patients, l’obtention d’une glycémie à jeun normale et une hémoglobine glyquée à 5,5 %.
Association fréquente avec d’autres maladies auto-immunes
Mais revenons à la patiente de 46 ans traitée au Massachusetts General Hospital. Au terme d’un bilan biologique poussé, un taux élevé d’anticorps anti-ribonucléoprotéine a été détecté. Ces anticorps, qui font partie de la famille des auto-anticorps anti-nucléaires, orientent vers le diagnostic de maladie auto-immune, en l’occurrence un lupus érythémateux systémique ou une connectivite mixte (maladie inflammatoire chronique).
Lourd traitement immunosuppresseur
La patiente a finalement été traitée par immunosuppresseurs, comportant notamment du rituximab et du cyclophosphamide. Ce dernier a provoqué une atteinte hépatique et a été remplacé par des immunoglobulines intraveineuses et l’azathioprine.
L’évolution a été marquée par des complications liées à l’immunosuppression, notamment par la réactivation d’une blastomycose (infection pulmonaire provoquée par un champignon), une candidose œsophagienne et une colite à Clostridioides difficile (anciennement Clostridium difficile), inflammation du côlon provoquant des diarrhées sévères. Ces infections ont entraîné de multiples interruptions du traitement immunosuppresseur.
Six mois après son transfert initial au Massachusetts General Hospital, cette femme reçoit toujours de l’insuline à haute dose et pèse 18 kg de plus. « La patiente continue de suivre un traitement immunomodulateur, avec surveillance de possibles effets secondaires, dans le but de parvenir à une rémission complète du syndrome de résistance à l’insuline de type B », concluent les auteurs.
Marc Gozlan (Suivez-moi sur X, Facebook, LinkedIn, Mastodon, Bluesky)