C’est l’histoire d’un jeune homme de 19 ans que sa mère amène aux urgences. Il indique aux médecins qu’il se lève trois à quatre fois par nuit pour uriner. Il a également perdu du poids et manque d’appétit. Il présente une soif excessive (polydipsie). Son poids est de 51,3 kg pour 1,65 m. Son indice de masse corporelle (IMC) est de 18,8 (normal).
Ce jeune patient allait bien jusqu’à ses 16 ans, âge où il a commencé à avoir des douleurs abdominales diffuses, des nausées et vomissements. Les mictions étaient également fréquentes (polyurie).
Sa glycémie est particulièrement élevée (7,8 g/L), de même que le taux d’hémoglobine glyquée qui est à 12,9 % (normale < 6 %). Le taux de créatinine, qui renseigne sur la fonction rénale, est de 1,45 mg/dL (valeur normale entre 0,6 et 1,50 mg/dL). Le diagnostic de diabète est établi. Le jeune homme est traité par insuline et hospitalisé.
Les trois jours suivants, il reçoit un schéma basal-bolus, qui associe une insuline basale (insuline lente) et une insuline apportée par un bolus, en l’occurrence une injection d’insuline rapide au moment des repas. Les douleurs abdominales, les nausées, les vomissements, la polyurie et la polydipsie disparaissent. Le 4e jour d’hospitalisation, la glycémie à jeun le matin tombe à 1,62 g/L et le taux de créatinine descend à 1,33 g/dL. Le patient regagne son domicile avec pour consigne de poursuivre son traitement par insuline. Un suivi est programmé pour qu’il soit suivi par un endocrinologue dans un autre hôpital.
Deux ans s’écoulent durant lesquels le patient suit son traitement insulinique. Des examens à la recherche d’auto-anticorps (GAD65, IA-2, anti-insuline) sont négatifs. On a détecté, lors d’un examen sanguin réalisé quatre ans plus tôt, la présence de peptide C, ce qui montre qu’il existe bien une sécrétion d’insuline. Par ailleurs, il n’existe pas de maladie auto-immune dans la famille du patient. Tous ces éléments sont peu en faveur d’un diabète de type 1 (DT1).
Détection d’anomalies du bilan hépatique deux ans après le diagnostic de diabète
Un an avant sa récente admission, le patient avait été adressé dans un service de gastroentérologie car les analyses biologiques sanguines avaient montré des taux élevés d’enzymes produites par le foie (transaminases ASAT/ALAT et phosphatases alcalines).
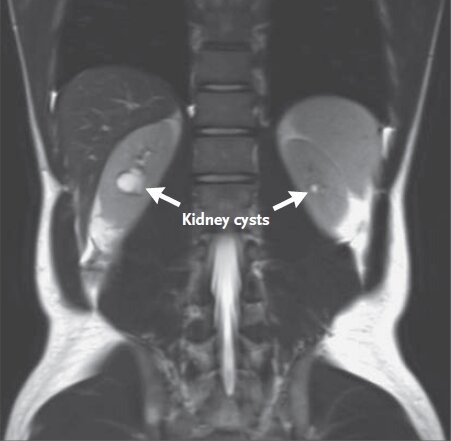
Une échographie de l’abdomen avait par ailleurs montré la présence de deux kystes, de un centimètre de diamètre, dans le rein droit. Une cholangiopancréatographie par résonance magnétique, application de l’IRM à l’étude du système biliaire et pancréatique, montre l’absence d’anomalies du foie, des voies biliaires et de la rate. En revanche, le pancréas est atrophique : la tête et la queue de cet organe étaient plus petits que la normale. En d’autres termes, on observe ce que l’on appelle une hypoplasie du pancréas.
Kystes multiples dans les deux reins
Enfin, cet examen d’imagerie détecte la présence de multiples kystes rénaux bilatéraux d’une taille comprise entre 2 et 18 mm.
Après ces examens radiologiques, le patient ne s’est pas rendu aux consultations de suivi programmées par les gastroentérologues. Il ne l’a fait qu’un an plus tard, une semaine avant son plus récent séjour à l’hôpital. Il ne présente alors pas de symptômes. Les images IRM sont inchangées par rapport aux précédentes.
Lors de son actuelle hospitalisation, les médecins observent que le patient se sent bien. Il suit son traitement par insuline. Il vit alors aux États-Unis, en Nouvelle-Angleterre, avec sa mère et trois frères et sœurs. Son père, obèse et hypertendu, avait un prédiabète.
Ce jeune homme, porteur de kystes dans les deux reins, est adressé dans le service de néphrologie. Son IMC est alors à 21,2 (il pèse alors 65,5 kg et mesure 1,76 cm). Il présente une légère protéinurie. Cette présence anormale de protéines dans les urines témoigne d’une atteinte rénale. Les analyses urinaires montrent également une glycosurie (présence de glucose dans les urines). L’échographie montre une structure hyperéchogène du parenchyme rénal et confirme la présence de nombreux kystes bilatéraux.
En résumé, ce jeune de 19 ans, atteint de diabète depuis l’âge de 16 ans, a des résultats anormaux de la fonction hépatique et est porteur de kystes rénaux. Il présente une histoire familiale de diabète, une protéinurie, la présence d’un pancréas atrophié et de reins hyperéchogènes à l’échographie. Comment ces signes cliniques et paracliniques disparates peuvent-ils être reliés à une pathologie ?
Les médecins, israéliens et américains, qui rapportent ce cas clinique complexe dans le numéro daté du 23 novembre 2023 du New England Journal of Medicine, notent que le diabète dont souffre leur patient n’est pas de type 1, dans la mesure où l’on ne retrouve pas les auto-anticorps associés à cette forme de diabète. Il présente très probablement un diabète imputable à un défaut sur un gène (diabète monogénique). Le fait qu’il y ait une histoire familiale de diabète va également dans ce sens. En outre, la présence de kystes rénaux depuis plus de trois mois et la protéinurie laissent penser que le patient présente une insuffisance rénale chronique.
Malgré le fait que ce patient présente un diabète, il est improbable que ses kystes rénaux soient en rapport avec une néphropathie diabétique. En effet, lorsque le rein est touché par la maladie diabétique, on n’observe généralement pas de kystes chez l’adulte jeune.
Les médecins du Centre médical Sheba (Ramat Gan, Israël) et du Massachusetts General Hospital (Boston) vont alors passer en revue la plupart des causes possibles d’une insuffisance rénale chronique (IRC) chez un adolescent, sachant que des centaines de gènes ont été identifiés comme pouvant être responsables d’une IRC. Une cause génétique est d’ailleurs impliquée dans environ 30 % des cas d’IRC diagnostiquée pendant l’adolescence. Surtout, la piste génétique est hautement privilégiée dans la mesure où ce jeune patient, porteur de kystes dans les reins, présente également des signes extra-rénaux (diabète, hypoplasie du pancréas, anomalies de la fonction hépatique).
Les signes cliniques observés chez ce patient ne sont pas typiques de ceux rencontrés en cas de polykystose rénale autosomique dominante (ADPKD), qui est la cause la plus fréquente de maladie rénale chronique chez l’adulte.
De même, la présentation clinique montre qu’il ne s’agit pas d’une polykystose rénale autosomique récessive (ARPKD), maladie kystique génétique rare touchant les reins et le foie.
Enfin, les médecins excluent que leur patient puisse être atteint de néphronophtise, pathologie génétique associant une atteinte rénale avec maladie kystique rénale évoluant vers l’insuffisance rénale terminale, mais associant de nombreux autres signes, notamment des atteintes oculaire et hépatique ainsi qu’un déficit intellectuel (retard mental).
Un dernier diagnostic est à évoquer, en l’occurrence une maladie génétique associée à une anomalie du gène HNF1B (Hepatocyte nuclear factor-1 β), situé sur le chromosome 17.
HNF1B, un gène qui joue un rôle important dans le développement embryonnaire
Ce gène appartient à la famille des gènes du développement. Il code pour ce que l’on appelle un facteur de transcription, protéine influençant l’expression d’un ou plusieurs gènes en se liant dans l’ADN à des séquences dites régulatrices. HNF1B est un gène codant pour un facteur de transcription impliqué dans le développement de l’appareil urogénital, du pancréas et du foie.
Ceci explique que la maladie associée au gène HNF1B se manifeste par des anomalies touchant plusieurs organes, ou puisse ne toucher que le rein, ou se traduire par des malformations de l’appareil urinaire. En effet, les signes cliniques de la maladie peuvent être variables d’un patient à l’autre au sein d’une même famille (hétérogénéité phénotypique intra-familiale).
Une maladie de présentation très variable
La variation du gène HNF1B provoque une maladie qui se manifeste de manière très diverse, allant de la présentation classique, associant kystes rénaux et diabète, à des symptômes cliniques moins spécifiques et parfois isolés tels qu’une malformation des voies urinaires ou une anomalie génitale.
Les kystes rénaux sont l’un des signes les plus fréquents de cette maladie génétique, souvent associés à des troubles de la fonction hépatique, comme ce qui a été observé chez le patient dont le cas est rapporté. Une hypoplasie du pancréas a également été décrite.
Cette pathologie, qui combine atteinte rénale et manifestations extrarénales, a été dénommée « kystes rénaux et diabète » (RCAD, renal cysts and diabetes). Elle est transmise sur le mode autosomique dominant.
Une portion du gène HNF1B peut manquer (délétion). Dans d’autres cas, sa séquence comporte une mutation ponctuelle. Une mutation peut subitement apparaître au sein d’une famille qui ne comptait pas de malades jusqu’alors. On parle de mutation de novo dans le cas où le patient est porteur d’une mutation absente chez ses parents. Chez plus de la moitié des cas, on détecte la présence de délétions hétérozygotes (perte d’un fragment sur une des deux copies du gène HNF1B).
Détecter la présence de ces délétions n’est pas chose facile et nécessite la mise en œuvre de techniques d’analyse génétique plus sophistiquées que le simple séquençage.
Les médecins ont donc effectué une analyse génétique qui a permis de diagnostiquer la présence d’un rare variant de HNF1B. Cette anomalie entraîne une perte totale de fonction de ce gène, ce qui fournit une preuve très forte en faveur de sa responsabilité dans la maladie observée chez ce jeune patient.
Jusqu’à présent, ce variant du gène HNF1B n’avait été décrit que chez trois autres malades, dont deux (non apparentés) présentaient une maladie rénale kystique et un autre chez lequel on suspectait un diabète d’origine génétique (MODY, maturity onset diabetes of the youth).
Ce n’est donc qu’après avoir éliminé les causes les plus fréquentes d’insuffisance rénale chronique chez leur jeune patient que les cliniciens ont suspecté une maladie associée au gène HNF1B et finalement identifié l’anomalie génétique très probablement responsable de sa maladie, qui associe kystes rénaux et diabète.
Maladie rénale et diabète MODY
Comme nous l’avons dit plus haut, on pouvait penser, au regard de l’absence d’auto-anticorps et de la détection du peptide C, que le diabète de ce jeune patient n’était probablement pas de type 1 (DT1). Le diagnostic est plutôt en faveur d’un diabète MODY, forme particulière de diabète d’origine génétique et qui est d’ailleurs le type le plus fréquent des diabètes monogéniques (ainsi dénommés car dus à l’anomalie d’un seul gène).
Le diabète MODY associé au gène HNF1B (anciennement dénommé MODY-5) peut être le premier symptôme manifeste de la maladie, avant même la survenue de l’atteinte rénale (néphropathie), à l’instar de ce qui a été observé chez le patient dont le cas est aujourd’hui rapporté.
La présence d’un peptide C détectable dans le sang indique que ce patient conserve une sécrétion d’insuline et peut donc sans doute être traité par des médicaments anti-diabétiques et se passer d’une insulinothérapie.
Suivi indispensable en gastroentérologie et néphrologie
Ce patient, qui présente des anomalies du bilan hépatique, continuera à être suivi par un gastroentérologue pour surveiller l’état de son système hépato-biliaire. De nombreux patients présentant la même pathologie restent toutefois asymptomatiques et ne reçoivent pas de traitement.
Ce jeune homme sera également régulièrement suivi par un néphrologue pour évaluer la progression de sa néphropathie qui peut se manifester par une hypertension artérielle, une augmentation de la protéinurie ou une aggravation de la dysfonction rénale.
Le traitement des manifestations rénales et extra-rénales dues à HNFIB est purement symptomatique.
Conseil génétique au patient et à la famille
Ce jeune patient pourrait être le premier membre de sa famille atteint de cette pathologie. Chez un patient sur deux, on ne trouve pas de cas antérieur de la maladie dans la famille.
La détection d’une maladie causée par le gène HNF1B chez un patient doit systématiquement entraîner des investigations pour rechercher la maladie chez ses parents, même si ceux-ci sont asymptomatiques.
Une information a été transmise par un généticien aux proches du jeune homme en vue de leur proposer un test génétique diagnostic. Il s’agit de rechercher la même mutation chez les deux parents du jeune homme, même si aucun d’eux ne présente de diabète ou de maladie rénale. En effet, tous les sujets porteurs d’une anomalie génétique du gène HNF1B ne développent pas la maladie (on parle de pénétrante incomplète). De plus, il existe une variabilité phénotypique familiale, tous les sujets d’une même famille ne présentant pas forcément les mêmes signes cliniques.
Un conseil génétique approprié doit également être apporté au patient car il existe un risque sur deux qu’il transmette la mutation à sa descendance.
Que retenir pratiquement de ce cas clinique ? Tout simplement que n’importe quel médecin doit savoir évoquer la maladie due à HNFI1B devant tout patient présentant des kystes rénaux et un diabète. L’interrogatoire de la famille est alors essentiel.
En cas de suspicion de cette maladie, dont on peut souligner qu’elle était inconnue il y a à peine une vingtaine d’années, il importe de consulter un médecin hospitalier spécialisé en endocrinologie ou en néphrologie afin d’établir le diagnostic au vu des signes cliniques et de l’analyse génétique.
Marc Gozlan (Suivez-moi sur X, Facebook, LinkedIn, Mastodon, Bluesky)