C’est l’histoire d’une mère de 36 ans et de ses deux jumeaux. Née de parents n’ayant pas de lien de consanguinité, cette femme a un hyperinsulinisme congénital néonatal.
Commençons par décrire cette maladie endocrinienne. L’hyperinsulinisme congénital est la cause la plus fréquente d’hypoglycémie chez le nouveau-né et le nourrisson. Cette pathologie est définie par la sécrétion inappropriée d’insuline par les cellules bêta des îlots de Langerhans du pancréas. Elle est responsable d’hypoglycémies sévères dont la prévention est essentielle pour prévenir le risque de séquelles cérébrales (épilepsie, retard psychomoteur). Le traitement de cette pathologie reste un défi pour les pédiatres.
L’incidence de l’hyperinsulinisme congénital est estimée entre 1 cas pour 40 000 naissances et 1 cas pour 2 500 naissances dans des régions où le taux de consanguinité est élevé.
Les symptômes apparaissent en général entre les premiers jours de vie et le 5e ou 6e mois. L’hypoglycémie peut entraîner un coma, voire le décès de l’enfant.
Deux formes histologiques : focale et diffuse
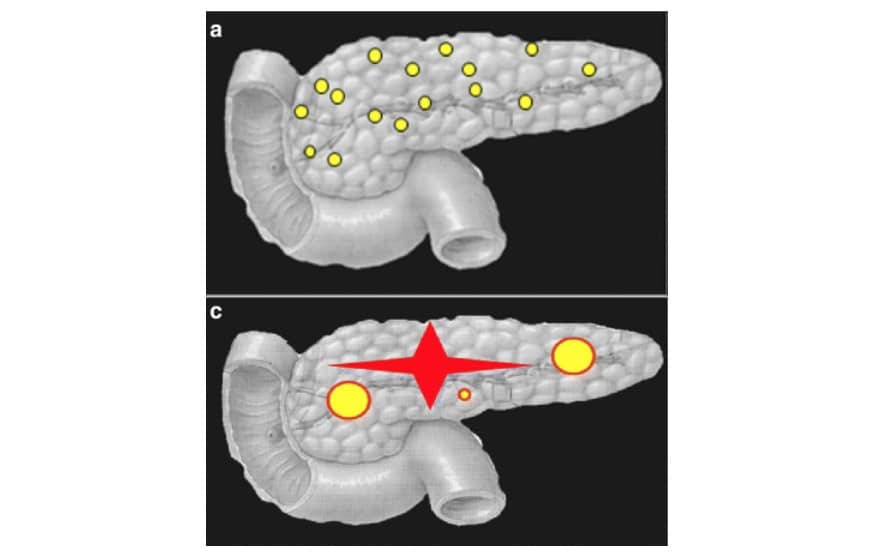
Il existe deux formes histologiques d’hyperinsulinisme congénital : la forme focale et la forme diffuse. La forme focale (30 % à 40 % des cas) est caractérisée par une hyperplasie pancréatique localisée des cellules bêta, autrement dit par un nombre excessif de ces cellules qui sont hyperfonctionnelles dans certains îlots. Le reste de la glande pancréatique est normal. Dans la forme diffuse (60 % à 70 % des cas), c’est l’ensemble de la glande qui est anormal, les îlots de Langerhans étant hyperactifs.
Les deux formes de la maladie ne peuvent être distinguées sur le plan clinique. L’imagerie médicale fonctionnelle, en l’occurrence par la tomographie par émission de positons avec injection de 6-fluoro-[18F]-L-dihydroxyphénylalanine (18F-DOPA), permet de localiser une possible lésion focale. Chez les enfants avec une forme focale, on observe une accumulation de 18F-DOPA, ce qui permet de localiser la lésion.
Dysfonctionnement du canal potassique ATP-dépendant des cellules bêta pancréatiques
Pour comprendre la suite, il convient de dire un mot sur le canal potassique de la cellule bêta, sécrétrice d’insuline. La membrane de ces cellules pancréatiques possède des canaux sélectifs pour le potassium : ce sont les canaux potassiques. Ils sont constitués des sous-unités Kir6.2 et SUR1. On parle de canal potassique car celui-ci forme un pore qui traverse la membrane plasmique de la cellule et laisse passer les ions potassium (K+).
Ce pore est composé de quatre sous-unités internes (Kir6.2), dont chacune possède un site de fixation pour l’ATP (molécule énergétique). Ce sont ces sous-unités qui permettent le passage des ions K+. La sous-unité Kir6.2 du canal potassique est codée par le gène KCNJ11. La synthèse de la sous-unité régulatrice SUR1 dépend, elle, du gène ABCC8, situé sur le bras court du chromosome 11 (région 11p15.1).
La plupart des patients atteints d’hyperinsulinisme congénital sont porteurs d’anomalies sur les gènes ABCC8 et KCNJ11. On connaît aujourd’hui une quinzaine de gènes impliqués dans cette pathologie.
Différences entre les formes focale et diffuse sur le plan génétique

Les formes focales sont dues à une mutation hétérozygote du gène ABCC8 ou du gène KCNJ11 (une seule copie du gène est mutée), qui est toujours héritée du père. S’y associe une seconde anomalie génétique (second-hit). Celle-ci peut être une perte d’allèle de la région chromosomique 11p15, d’origine maternelle et limitée aux cellules pancréatiques de la lésion focale. Il y a donc transmission d’une mutation par le père associée à une perte d’expression de la région correspondante du chromosome maternel. Chez certains patients, cette seconde anomalie peut résulter d’un mode de transmission inhabituel de la région 11p15 d’origine paternelle (isodisomie uniparentale paternelle résultant en une duplication de l’allèle paternel).
Dans les formes diffuses, différents mécanismes de transmission et plusieurs gènes peuvent être impliqués. Dans ces cas, on observe en effet une grande hétérogénéité génétique. Les nouveau-nés présentant une forme diffuse sévère sont porteurs d’un variant dominant du gène ABCC8, ou d’une mutation homozygote (les deux copies du gène ABCC8 ou KCNJ11) sont mutées), ou encore de deux mutations récessives hétérozygotes du gène ABCC8 (mutations composites).
Lorsque le traitement médical s’avère inefficace dans la forme diffuse, une pancréatectomie subtotale (ablation d’un volume important de la glande pancréatique) reste l’unique recours. Les pancréatectomies subtotales réalisées pour un hyperinsulinisme congénital diffus sont fréquemment suivies de fluctuations du taux de glucose plasmatique (hypoglycémies ou hyperglycémies postopératoires) et à long terme d’un diabète insulinodépendant.
Revenons à la patiente de 36 ans dont le cas a été rapporté le 17 juin 2024 dans la revue Frontiers in Endocrinology par Daniela Telehuz, Olivier Dupuy, Jean-Baptiste Arnoux et leurs collègues des hôpitaux Saint-Joseph et Necker-Enfants malades (Paris). Cette femme a été diagnostiquée comme atteinte d’un hyperinsulinisme congénital néonatal. Elle ne répondait pas au traitement par diazoxide (agoniste du canal KATP qui se fixe sur sa sous-unité SUR1 et inhibe la sécrétion d’insuline).
Pancréatectomie subtotale à l’âge de trois semaines
À l’âge de trois semaines, elle a subi une pancréatectomie subtotale qui l’a guérie de son hypoglycémie. C’était le traitement standard préconisé en 1985. À l’époque, on ne connaissait pas encore l’existence des formes focales, qui sont curables par pancréatectomie partielle (ablation d’une partie du pancréas). Les patients opérés d’une forme focale d’hyperinsulinisme congénital par pancréatectomie limitée sont en effet définitivement guéris.
À l’âge de deux ans, la fillette a eu une analyse histologique du pancréas, qui a alors montré qu’elle présentait une lésion focale. L’analyse génétique n’était pas disponible à cette époque.
À neuf ans, cette jeune patiente a développé un diabète insulinodépendant. La pancréatectomie subtotale est en effet suivie d’un risque important de diabète à court ou long terme. Elle est aujourd’hui traitée par pompe à insuline.
Désir de grossesse et conseil génétique
En 2016, cette femme a un désir de grossesse et en parle à son endocrinologue qui l’adresse à un médecin généticien pour avoir un conseil génétique. Au vu de l’histoire médicale, un test génétique est réalisé, qui révèle la présence d’une mutation hétérozygote du gène ABCC8 héritée de son père.
Chez cette patiente née en 1985, le diagnostic moléculaire précis de son hyperinsulinisme congénital a donc été confirmé plus de deux décennies après le début de sa maladie. Il est à noter que la mutation non-sens du gène ABBC8 (conduisant à la formation d’un message d’arrêt de la synthèse de la protéine) dont cette patiente est porteuse n’avait jamais été décrit avant 2020.
Considérant que l’histologie du pancréas indiquait une forme focale, le variant du gène ABBC8 est considéré comme ayant été transmis sur le mode récessif. Le généticien en conclut que le risque pour cette femme d’avoir un enfant atteint d’hyperinsulinisme congénital est très faible (1 sur 600). En outre, du fait de l’absence de consanguinité dans le couple, il n’est pas jugé nécessaire que le père se soumette à un test génétique.
Une grossesse débute en 2020. Une rupture prématurée des membranes survient à l’âge gestationnel de 26 semaines. Une césarienne est réalisée en urgence à 28 semaines et 6 jours.
Naissance de garçons jumeaux atteints d’hyperinsulinisme
La patiente donne naissance à deux garçons jumeaux. Ils souffrent tous deux d’hypoglycémie persistante sévère. Neuf jours plus tard (J9), les analyses sanguines révèlent un hyperinsulinisme chez les deux nourrissons.
L’objectif du traitement consiste à prévenir les lésions du cerveau liées à l’hypoglycémie. Une prise en charge précoce permet de prévenir le risque de séquelles cérébrales en cas d’hypoglycémies sévères ou prolongées.
Un traitement par diazoxide est entrepris à J51. Il est interrompu à J69 pour le second jumeau et à J72 pour le premier, car il s’avère inefficace et provoque des effets secondaires hémodynamiques (hypertension pulmonaire et œdème aigu du poumon).
Les médecins décident alors d’administrer d’abord par perfusion, puis par voie sous-cutanée, de l’octréotide (médicament qui inhibe la sécrétion d’insuline par le canal KATP). La persistance d’épisodes hypoglycémiques malgré l’administration de doses maximales d’octréotide conduit à changer de traitement. Celui-ci est remplacé par le pasiréotide, médicament censé avoir un effet hyperglycémiant plus important. Malgré ce nouveau traitement agressif, plusieurs épisodes d’hypoglycémie sont encore observés.
Examens génétiques révélateurs
Des analyses génétiques sont réalisées chez les deux nourrissons, révélant deux génotypes identiques. Leur mère leur a transmis son variant du gène ABCC8 (mutation non-sens). En outre, de façon inattendue, les analyses indiquent que le père leur a transmis un autre variant du gène ABCC8 (mutation troncative, c’est-à-dire conduisant à la synthèse d’une protéine plus courte et non fonctionnelle).
Un père, asymptomatique, porteur d’un variant ABCC8
Ces jumeaux ont donc hérité d’une anomalie génétique transmise par chaque parent. Il s’avère que le père, asymptomatique, est porteur sain d’une mutation récessive du gène ABCC8. Il est à noter que ce variant paternel n’avait pas été répertorié jusqu’à présent dans la base de données génétiques Human Gene Mutation Database (HGMD).
Les deux jumeaux, du fait qu’ils possèdent deux variants récessifs pathogènes du gène ABCC8, ont été diagnostiqués comme étant atteints d’une forme diffuse d’hyperinsulinisme congénital.
Des jumeaux avec une forme différente d’hyperinsulinisme congénital de celle de leur mère
Ce cas est exceptionnel dans la mesure où une mère atteinte d’une forme focale d’hyperinsulinisme congénital a transmis à ses deux jumeaux la forme diffuse de cette maladie.
Les deux nourrissons ont quitté l’hôpital à l’âge de cinq mois. Ils ont reçu une nutrition entérale en continu à domicile, les nutriments étant administrés directement dans l’estomac.
Le traitement par pasiréotide est poursuivi à domicile par voie injectable sous-cutanée via un système de pompe. Lorsque les enfants ont 12 mois, le pasiréotide est remplacé par une formulation à action longue (à raison d’une injection toutes les quatre semaines). Un traitement par diazoxide est de nouveau entrepris. Il est bien toléré jusqu’à ce jour. Grâce à ce traitement, la survenue d’épisodes hypoglycémiques est rare et les médecins n’envisagent pas d’avoir recours à une pancréatectomie subtotale.
À l’âge de 3 ans, les deux jumeaux sont toujours lourdement traités, avec notamment diazoxide en injection intramusculaire, pasiréotide injectable, alimentation entérale la nuit. Fort heureusement, leur croissance est normale. Ils s’apprêtent à aller à l’école dans quelques mois. Ils n’ont eu aucun effet secondaire de leur traitement, si ce n’est une légère hyperpilosité, effet indésirable connu du diazoxide.
Les parents ont été informés du risque élevé de récidive d’hyperinsulinisme congénital lors d’une nouvelle grossesse, avec un risque de 25 % d’une forme diffuse, mais aussi un risque estimé à 1/1200 d’une forme focale. En raison de la gravité de la maladie dans cette famille, il va sans dire qu’un diagnostic prénatal serait proposé à ce couple.
Un scénario a priori improbable en l’absence de consanguinité
La situation décrite par les cliniciens parisiens correspond, en l’absence de parents consanguins, à un scénario a priori improbable. Selon eux, « ce scénario particulier, survenu en dehors de consanguinité, est un événement rare qui pose un défi important pour la communauté médicale et incite à réévaluer le contexte des tests génétiques au-delà de la consanguinité en même temps qu’il soulève des questions pertinentes quant à sa mise en œuvre ». Et de conclure en estimant qu’il est « impératif de considérer les tests génétiques comme un outil de prévention et de prise en charge de l’hyperinsulinisme néonatal en dehors du contexte de consanguinité lorsqu’un des parents est déjà connu comme étant porteur d’une mutation ABCC8 ».
Marc Gozlan